Кариотип при синдроме эдвардса
Синдром Эдвардса
Синдром Эдвардса– хромосомное заболевание, обусловленное трисомией по 18-ой хромосоме и сопровождающееся множественными пороками развития. Для синдрома Эдвардса характерны своеобразные фенотипические признаки (долихоцефалическая форма черепа, микрофтальмия, недоразвитие ушных раковин, микроретрогнатия и др.), аномалии опорно-двигательной, сердечно-сосудистой, пищеварительной, мочеполовой системы, ЦНС. Синдром Эдвардса может быть диагностирован на этапе беременности (УЗИ-скрининг, инвазивная пренатальная диагностика) либо уже после рождения ребенка на основании внешних признаков и цитогенетического исследования. Дети с синдромом Эдвардса нуждаются в симптоматическом лечении и хорошем уходе.
Общие сведения
Синдром Эдвардса – количественная хромосомная аберрация, при которой имеет место частичная или полная трисомия по 18 аутосоме. Синдром получил название по имени генетика J. Edwards, подробно описавшего заболевание в 1960 г. и выделившего свыше 130 характерных для данной патологии симптоматических дефектов. Синдром Эдвардса – второе по распространенности хромосомное заболевание после синдрома Дауна; частота рождения детей с синдромом Эдвардса составляет 1:5000-7000. Примерно три четверти всех больных синдромом Эдвардса – девочки; предполагается, что большая часть беременностей плодом мужского пола заканчивается внутриутробной гибелью и самопроизвольным абортом.
Причины синдрома Эдвардса
Развитие синдрома Эдвардса объясняется хромосомными нарушениями, происходящими на стадии гаметогенеза (овогенеза или сперматогенеза) либо дробления зиготы и приводящими к увеличению числа хромосом 18-й пары. В 80-90% случаев цитогенетические варианты синдрома Эдвардса представлены простой трисомией 18, реже – мозаичной формой или несбалансированными перестройками (транслокациями).
Причиной полной трисомии служит мейотическое нерасхождение хромосом. Практически во всех случаях лишняя хромосома является материнской по происхождению. Этот вариант синдрома Эдвардса является наиболее тяжелым по своим проявлениям и неблагоприятным в плане прогноза. Возникновение мозаицизма связано с нерасхождением хромосом на ранней стадии дробления зиготы. В этом случае лишнюю хромосому будут содержать не все клетки плода, а лишь их часть. Транслокация – присоединение части 18-ой хромосомы к другой паре может произойти как в процессе созревания гамет, так и после оплодотворения. При этом клетки организма содержат две гомологичные 18-е хромосомы и ее дополнительную часть, прикрепленную к другой хромосоме.
Как и в случае с синдромом Дауна, возраст матери является наиболее значимым риск-фактором рождения ребенка с синдромом Эдвардса. В редких случаях у родителей может выявляться носительство сбалансированной транслокации.
Симптомы синдрома Эдвардса
Во время беременности наблюдается многоводие, слабая активность плода, маленькая плацента, единственная пупочная артерия. Ребенок с синдромом Эдвардса рождается с низкой массой тела (около 2170 г) и пренатальной гипотрофией при доношенной или даже переношенной беременности. У части детей определяется состояние асфиксии при рождении.
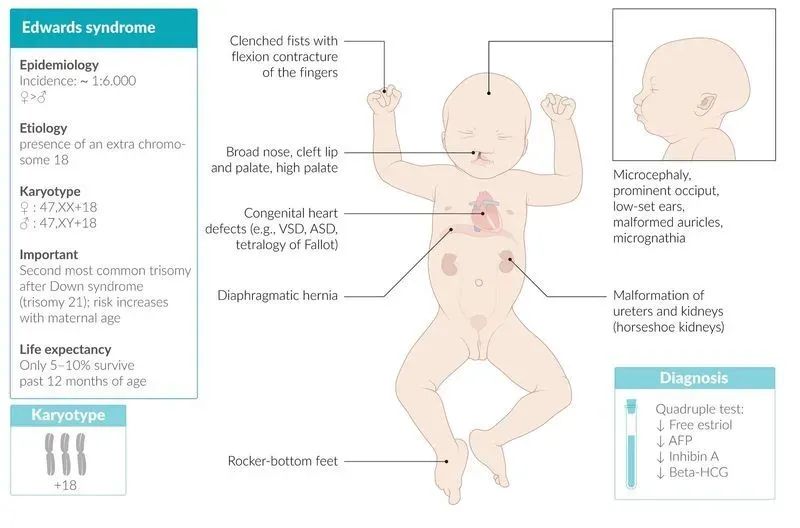
У новорожденных с синдромом Эдвардса имеются характерные фенотипические признаки, позволяющие предположить данную хромосомную патологию. В первую очередь обращает на себя внимание долихоцефалическая форма черепа с преобладанием продольного размера над поперечным, низкий лоб, выступающий затылок, микрогнатия, маленький рот, микрофтальмия. У детей с синдромом Эдвардса часто встречаются расщелины верхней губы и нёба, эпикант, птоз, экзофтальм, косоглазие, короткая шея с избыточной кожной складкой. Типичные деформации ушных раковин включают маленькие мочки, отсутствие козелков, узкие слуховые проходы, низкое расположение ушей.
Внешний облик детей дополняется характерными для синдрома Эдвардса деформациями скелета – скрещенными пальцами кистей, укороченной грудиной, аномалиями ребер, врожденным вывихом бедра, косолапостью, «стопой-качалкой», синдактилией стоп и пр. У многих детей имеются гемангиомы и папилломы кожи.
При синдроме Эдвардса имеются множественные тяжелые аномалии со стороны практически всех систем организма. Врожденные пороки сердца могут быть представлены дефектами межжелудочковой и межпредсердной перегородок, коарктацией аорты, транспозицией магистральных сосудов, дисплазией клапанов, тетрадой Фалло, аномальным дренажом легочных вен, декстракардией и др. При синдроме Эдвардса может выявляться патология развития желудочно-кишечного тракта: диафрагмальные, пупочные и паховые грыжи, дивертикул Меккеля, трахеопищеводные свищи, пилоростеноз, атрезия подвздошной кишки и ануса. Наиболее частыми аномалиями мочеполовой системы у детей с синдромом Эдвардса служат подковообразная почка, гидронефроз, дивертикулы мочевого пузыря, гипоспадия и крипторхизм (у мальчиков), двурогая матка, внутриматочная перегородка и гипертрофия клитора (у девочек).
Пороки развития центральной нервной системы характеризуются наличием микроцефалии, менингомиелоцеле, гидроцефалии, аномалии Арнольда-Киари, кист арахноидального сплетения, гипоплазии мозжечка и мозолистого тела. У всех выживших детей с синдромом Эдвардса имеются интеллектуальные нарушения – олигофрения в степени глубокой имбецильности или идиотии.
Новорожденные с синдромом Эдвардса испытывают трудности с сосанием, глотанием и дыханием, из-за чего им требуется зондовое питание или длительная ИВЛ. Дети с синдромом Эдвардса, как правило, погибают на первом году жизни из-за тяжелых врожденных пороков развития и связанных с ними осложнений (сердечно-сосудистой и дыхательной недостаточности, пневмонии, кишечной непроходимости и т. д.).
Диагностика синдрома Эдвардса
Важнейшей задачей диагностики служит антенатальное выявление синдрома Эдвардса у плода, поскольку данная патология является медицинским показанием для искусственного прерывания беременности. Заподозрить наличие синдрома Эдвардса можно в процессе УЗИ плода и допплерографии маточно-плацентарного кровотока по косвенным признакам (множественным аномалиям развития плода, агенезии пупочной артерии, малой величине плаценты, многоводию и пр.).
Наибольшую диагностическую значимость имеет стандартный пренатальный скрининг, включающий анализ крови на сывороточные маркеры: βХГЧ и PAPP на 11-13 неделе беременности; βХГЧ, альфа-фетопротеин и свободный эстриола на 20-24 неделе гестации.
При оценке степени риска рождения ребенка с синдромом Эдвардса учитываются данные биохимического и ультразвукового скрининга, срок беременности, возраст и масса тела женщины. Беременным, попадающим в группу высокого риска, предлагается проведение инвазивной дородовой диагностики (биопсии хориона, амниоцентеза, кордоцентеза) с последующим кариотипированием плода.
В случае рождения живого ребенка с синдромом Эдвардса необходимо как можно более раннее всестороннее обследование, направленное на выявление тяжелых пороков развития. Новорожденный с синдромом Эдвардса должен быть осмотрен неонатологом, детским кардиологом, детским неврологом, детским хирургом, детским ортопедом, детским урологом и др. Наиболее важными диагностическими исследованиями, которые должны быть выполнены ребенку с синдромом Эдвардса в первые часы жизни, служат эхокардиография, УЗИ органов брюшной полости и УЗИ почек.
Лечение синдрома Эдвардса
Поскольку в большинстве случаев аномалии развития оказываются несовместимыми с жизнью, лечение детей с синдромом Эдвардса сводится к оказанию симптоматической помощи, направленной на поддержание физиологических функций, продление жизни и улучшение ее качества. Хирургическая коррекция врожденных пороков, как правило, является рискованной и неоправданной.
Поскольку дети с синдромом Эдвардса ослаблены и подвержены частой заболеваемости инфекциями мочевыводящих путей, средним отитом, конъюнктивитом, синуситами, пневмониями и пр., они нуждаются в тщательно организованном уходе, полноценном питании, регулярном наблюдении со стороны педиатра.
Прогноз и профилактика синдрома Эдвардса
Во всех случаях прогноз при синдроме Эдвардса крайне неблагоприятный: в среднем мальчики живут 2-3 месяца, девочки – 10 месяцев. До 1 года доживает лишь 10% больных, до 10 лет – не более 1%. Относительно благоприятные шансы в отношении выживания имеют дети с мозаичной формой синдрома Эдвардса.
Свечи от цервицита воспаление шейки маткиРиск рождения ребенка с синдром Эдвардса теоретически существует в любой супружеской паре; известно, что такая вероятность выше у возрастных родителей (для женщин старше 45 лет – 0,7%). С целью своевременного выявления хромосомной патологии у плода не следует пренебрегать антенатальным скринингом, входящим в программу введения беременности.
Синдром Эдвардса
Синдром Эдвардса – генетическая патология, которая характеризуется трисомией (дублированием) 18 хромосомы и сопровождается рядом пороков развития.
| МКБ-10 | Q91 |
|---|---|
| МКБ-9 | 758.2 |
Содержание
Общая информация
Трисомия по 18 хромосоме (синдром Эдвардса) – ненаследственное заболевание, мутация генов происходит случайно. Патология встречается в 1 случае из 3000 зачатий и 6000-7000 рождений. У девочек она диагностируется чаще, чем у мальчиков в 3 раза.
Риск синдрома Эдвардса повышается с возрастом матери – после 45 лет он составляет 0,7%. Болезнь негативно отражается на течении беременности: отмечается многоводие, недостаточная активность плода и роды позже срока (на 42-43 неделе).
60% детей с синдромом Эдвардса умирают в период внутриутробного развития. Продолжительность жизни 2-3 месяца – для мальчиков, 10 месяцев – для девочек. Смерть наступает из-за серьезных нарушений в работе внутренних органов.
Причины
Причина синдрома Эдвардса – присутствие в наборе хромосом (кариотипе) «лишней» 18 хромосомы. В норме зигота (яйцеклетка после оплодотворения) должна иметь 23 пары хромосом, полученных в результате слияния женской и мужской гамет (половых клеток). Под влиянием не установленных наукой факторов происходит дублирование генетического материала. У зиготы вместо двух 18 хромосом появляется три: материнская, отцовская и дополнительная.
Мутация может осуществиться как до оплодотворения, так и после него – на этапе деления яйцеклетки. Кариотип при синдроме Эдвардса имеет три варианта:
- полная трисомия (95% случаев) – три 18 хромосомы в каждой клетке плода;
- транслокационная (2%) – две 18 хромосомы и часть третьей, добавившаяся к другой хромосоме;
- мозаичная (3%) – три 18 хромосомы в некоторых клетках будущего ребенка.
Первая форма трисомии синдрома Эдвардса характеризуется более сложным течением, чем две другие.
Симптомы
Симптомы синдрома Эдвардса варьируются в зависимости от условий развития эмбриона и формы трисомии. В большинстве случаев имеют место следующие внешние признаки:
- Масса тела при рождении – 2,1-2,2 кг.
- Искажение черепа – маленький размер, долихоцефалическая (удлиненная к затылку) форма, гидроцефалия.
- Деформация лицевых костей – узкий лоб, широкий затылок, недоразвитие нижней челюсти, маленький рот, высокое небо, нарушенный прикус, узкие глазные щели, углубленная переносица, суженный нос.
- «Заячья губа», «волчья пасть».
- Укороченная шея с кожной складкой.
- Низко посаженные уши неправильной формы – без козелка, вытянутые горизонтально, с узким слуховым ходом.
Синдром Эдвардса характеризуется значительным изменением опорно-двигательного аппарата. Обычно наблюдаются такие нарушения, как:
- Дисфункция суставов – они не могут нормально сгибаться и разгибаться.
- Дисплазия стоп – провисание свода с одновременным выступанием пятки («стопа-качалка»).
- Подвижность тазобедренных суставов.
- Расширение и укорочение грудной клетки
- Увеличение количеств дуг палацев рук, отсутствие сгибательной складки.
- Сращивание пальцев стопы, появление перепонок между ними.
При синдроме Эдвардса отмечаются множественные аномалии в работе внутренних органов, в частности:
- Пороки сердца – незакрытый артериальный проток, дефект перегородки между желудочками и так далее.
- Эндокринные проблемы, под влияние которых замедляется рост и недостаточно формируется подкожная клетчатка.
- Гипотония мышц.
- Аномалии пищеварительной системы – гастроэзофагеальныйрефлюкс, слабость сосательного рефлекса, атрезия некоторых участков ЖКТ, дивертикулы.
- Мочеполовые патологии – крипторхизм, недоразвитость яичников, деформация почек, удвоение мочеточников.
- Нарушение зрения.
Ребенок с синдромом Эдвардса
Каждый из признаков синдрома Эдвардса встречаются в 70-95% случаев. Абсолютно у всех детей есть проблемы с развитием головного мозга – умственная отсталость (вплоть до имбецильности).
Диагностика
Диагностику синдрома Эдвардса можно осуществить во время беременности Наиболее доступной методикой является ультразвуковое исследование.
Косвенные признаки синдрома Эдвардса на УЗИ на раннем этапе (12 недель):
- большой объем околоплодной жидкости;
- снижение частоты сердцебиения;
- брюшная грыжа;
- отсутствие носовых костей;
- 2 артерии в пуповине;
- кисты сосудистых сплетений.
На поздних сроках (третий триместр беременности) синдром Эдвардса на УЗИ может проявляться такими симптомами, как:
- аномалии костей головы и мягких тканей;
- пороки опорно-двигательного аппарата, сердца, сосудов, мочеполовой системы и так далее.
 УЗИ – один из методов, позволяющих предположить синдром Эдвардса
УЗИ – один из методов, позволяющих предположить синдром Эдвардса
Более точным способом диагностики является биохимический анализ крови на 11-13 неделе беременности. Исследование предполагает определение уровня плазменного протеина А (РАРР-А) и хорионического гормона (β-ХГЧ). Затем полученные результаты соотносят с возрастом будущей мамы и сроком вынашивания ребенка. При отклонении показателей от нормы ставят высокий риск синдрома Эдвардса. Диагноз на этом этапе не определяется.
Затем осуществляется определение статуса плода с помощью различных методик:
Инвазивные способы выявления синдрома Эдвардса:
- Биопсия ворсин хориона (БВХ) – исследование небольшого образца плаценты. Доступна на сроке 8-12 недель.
- Амниоцентез – проникновение через амниотическую оболочку и взятие пробы околоплодных вод. Делается на сроке 14-18 недель.
- Кордоцентез – забор пуповинной крови. Проводится в 20 недель.
Все три метода дают возможность определить кариотип ребенка и подтвердить либо опровергнуть диагноз. Их недостаток – риск инфицирования плода и прерывания беременности.
К неинвазивным методикам относится выделение ДНК ребенка из крови матери.
Точность генетического тестирования очень высока: синдром Эдвардса подтверждается часто – в 90% случаев. Сложнее всего установить мозаичную форму заболевания: в проверяемый образец могут не попасть клетки с измененным кариотипом.
После рождения ребенка патология диагностируется на основании внешнего осмотра и кариотипирования – исследования крови с целью определения хромосомного набора.
Лечение
Причины возникновения синдрома Эдвардса заключаются в генетической мутации, потому избавиться от этого заболевания невозможно ни до, ни после рождения.
Лечение синдрома Эдвардса сводится к максимальному устранению аномалий строения. Но из-за высокой смертности в первые месяцы жизни и наличия множественных нарушений в работе всех систем хирургическое вмешательство в большинстве случаев оказывается нецелесообразным. Как правило, помощь ребенку с синдромом Эдвардса заключается в:
- восстановлении проходимости пищевода и кишечника;
- кормлении через зонд либо применении специальных смесей;
- использовании слабительных и пеногасителей;
- создании стерильных условий, поскольку дети с синдромом Эдвардса крайне подвержены различным инфекционным заболеваниям.
При относительно легкой форме патологии после достижения ребенком 2-3 летнего возраста становится возможным проведение операций по устранению косметических дефектов («заячьей губы», сросшихся пальцев), а также различных аномалий внутренних органов (порока сердца, грыж и так далее). Кроме того, требуется постоянная коррекция задержки психического развития с помощью обучающих методик.
Лечение миомы маткиСиндром Эдвардса увеличивает риск возникновения ряда заболеваний, среди которых:
- рак почек (опухоль Вильмса);
- пневмония;
- конъюнктивит;
- мочеполовые инфекции;
- фронтит, отит;
- апноэ по ночам;
- сердечно-сосудистые патологии так далее.
Пациенты с трисомией 18 хромосомы нуждаются в регулярных медицинских обследованиях.
Прогноз и профилактика
Синдром Эдвардса характеризуется неблагоприятным прогнозом: 60% больных умирают до трехмесячного возраста, еще 30-35% – до года. Выжившие 5-10% детей страдают от умственной отсталости и различных заболеваний, связанных с аномалиями строения.
При легкой форме мозаичного синдрома Эдвардса психическое и физическое развитие детей можно корректировать: научить их общаться с ограниченным кругом людей, привить некоторые навыки самообслуживания.
Поскольку патология относится к случайным хромосомным мутациям, предупредить его возникновение невозможно.
Синдром Эдвардса
Синдром Эдвардса — хромосомное заболевание, характеризуется комплексом множественных пороков развития и трисомией 18 хромосомы. Описан в 1960 году Джоном Эдвардсом. Популяционная частота примерно 1:3000 в США, и 1:5000 в мире на 2016 год. Дети с трисомией в 18 чаще рождаются у пожилых матерей, взаимосвязь с возрастом матери менее выражена, чем в случаях трисомии хромосомы 21 и 13. Для женщин старше 45 лет риск родить больного ребёнка составляет 0,7 %.
Синдром Эдвардса – количественная хромосомная аберрация, при которой имеет место частичная или полная трисомия по 18 аутосоме. Синдром получил название по имени генетика J. Edwards, подробно описавшего заболевание в 1960 г. и выделившего свыше 130 характерных для данной патологии симптоматических дефектов. Синдром Эдвардса – второе по распространенности хромосомное заболевание после синдрома Дауна; частота рождения детей с синдромом Эдвардса составляет 1:5000-7000. Примерно три четверти всех больных синдромом Эдвардса – девочки; предполагается, что большая часть беременностей плодом мужского пола заканчивается внутриутробной гибелью и самопроизвольным абортом.
Причина заболевания
Если синдром Эдвардса (фото больных детей по этическим соображениям не будут размещены) был диагностирован во время беременности, то чаще всего последняя заканчивается выкидышем или мертворождением. Увы, проявление болезни у плода предотвратить сегодня нельзя. Также в современности не выяснены четкие причины этого генетического заболевания, отчего и профилактические меры против развития его у будущих детей сформировать нельзя. Однако специалисты определили факторы риска:
- Неблагоприятные экологические условия.
- Воздействие радиации, токсических, химических веществ на родителей.
- Пристрастие к алкоголю и табаку.
- Наследственность.
- Прием определенных медикаментов.
- Инцест, кровное родство родителей.
- Возраст будущей матери. Если женщине старше 35 лет, то это считается причиной синдрома Эдвардса у ребенка, равно как и иных хромосомных заболеваний.
Симптомы
Такое отклонение, как синдром Эдвардса, характеризуют множественные симптомы, но спутать с другими патологиями недуг невозможно. Первые симптомы отклонения у малыша можно обнаружить на УЗИ, когда он ещё находится в утробе матери, но если все же малыш родился с данным заболеванием, его симптомы будут очевидны:
- искажение лица;
- наличие заячьей губы;
- непропорционально маленькая голова или дефекты её строения;
- низкий вес малыша при рождении;
- наличие волчьей пасти;
- дефекты челюстей, слишком маленький рот, из-за чего часто затруднено кормление.
Другие симптомы заболевания, которые присущи детям с этой врождённой аномалией, это перепонки между пальцами, сжатые в кулаки пальцы на руках (пальцы при этом неравномерно согнуты), врождённая косолапость, слишком низко расположенные уши. Внешний вид такого ребёнка очень странный. Но кроме внешних признаков, есть и внутренние нарушения, которые, чаще всего, становятся причинно смерти детей.
Симптомы нарушений функционирования органов и систем при такой патологии, как синдром Эдвардса, следующие:
- отставание в физическом и умственном планах;
- наличие паховых и пупочных грыж;
- врождённые пороки сердечно-сосудистой системы;
- почка в виде подковы, удвоенные мочеточники и другие патологии развития мочеполовой системы (гигантский клитор у девочек, крипторхизм у мальчиков и т. д.);
- атрофия мышц, из-за чего у малыша развивается выраженный сколиоз и другие патологии костно-мышечной системы;
- косоглазие;
- нарушения работы пищеварительной системы вследствие недоразвитости её органов.
Диагностика патологии
Врач, оценивает степень риска рождения малыша с хромосомным заболеванием, учитывая данные УЗИ и биохимического анализа, срок беременности, массу тела и возраст женщины. Тем беременным, которые входят в группу риска, предлагается проведение инвазивной дородовой диагностики с последующим осуществлением кариотипированием плода. После рождения ребенка с синдромом следует максимально раннее детальное обследование, цель которого – выявление пороков развития малыша. Диагностика синдрома Эдвардса включает:
- эхокардиографию;
- УЗИ почек и органов брюшной полости;
- обследование неонатолога, невролога, кардиолога, ортопеда, уролога;
- осмотр детским хирургом.
Лечение заболевания
Терапия рассмотренной мутации направлена на смягчение ее симптомов и облегчение жизни младенца. Излечить синдром Эдвардса и обеспечить ребенку полноценное развитие нельзя. Стандартные медицинские мероприятия помогают:
- восстановить прохождение пищи при атрезии анального отверстия или кишечника;
- организовать кормление через зонд на фоне отсутствия сосательного и глотательного рефлексов;
- стабилизировать функционирование сердечно-сосудистой системы;
- нормализовать отток мочи.
Часто синдром Эдвардса новорожденных дополнительно требует применения противовоспалительных, антибактериальных, гормональных и других сильнодействующих препаратов. Это необходимо для своевременной интенсивной терапии всех сопутствующих заболеваний, которые он провоцирует:
- опухоль Вильмса;
- конъюнктивит;
- пневмония;
- средний отит;
- легочная гипертензия;
- синусит;
- мочеполовые инфекции;
- фронтит;
- высокое артериальное давление и другое.
Прогнозы
Для большинства детей, рожденных с заболеванием Эдвардса, дальнейший прогноз развития болезни неблагоприятен — те из них, которые не умирают до года и доживают до относительно взрослого возраста, имеют явное умственное торможение в развитии, не могут за собой ухаживать, за ними нужен постоянный уход и контроль. Но они понимают, когда с ними ласково обращаются, утешают, играют. Больные синдромом Эдвардса могут самостоятельно кушать, улыбаться и понемногу учиться всевозможным полезным бытовым навыкам.
То, что при этой болезни у больных имеется целый «букет» неправильно развитых и неработающих органов, приводит к высокому проценту детской смертности. Если по результатам обследования в первые месяцы беременности женщину относят к группе риска по данному заболеванию плода — обычно медики дают рекомендации к искусственному прерыванию беременности. Но окончательное решение принимает будущая мать. Часто женщина не решается прервать беременность, надеясь на ошибку медиков и благоприятный исход. К сожалению, эффективные способы борьбы с проявлением синдрома Эдвардса на данное время не найдены.
Синдром Эдвардса: клинические проявления и возможности терапии
Причины развития
Основная причина развития заболевания — изменение хромосомного набора. Трисомия 18 встречается чаще всего. В некоторых случаях у пациентов обнаруживают мозаичный вариант патологии, сопровождающийся мутациями в конкретных генах.
Увеличение количества 18 хромосомы возникает в результате ее нерасхождения между клетками при мейозе (процесс деления половых клеток). Нарушения возникают в яйцеклетках. У ребенка развивается тяжелая форма заболевания. Легкие варианты болезни связаны с мозаицизмом. Это ситуация, при которой часть хромосомного материала переносится, приводя к нарушению работы генов.
Соли мочи ураты что этоВ развитии хромосомных патологий важным факторов является возраст женщины. Если беременность возникла после 30 лет, то риск возникновения трисомии увеличивается в несколько раз.
Клинические проявления
Первые изменения при синдроме Эдвардса отмечаются в период беременности. При обследовании женщины выявляют многоводие, уменьшение размеров плаценты и наличие только одной пупочной артерии (в норме – 2 артерии). Масса детей при рождении меньше нормы – до 2100 г и менее, что связано с общей гипотрофией.
У детей с патологией выражены фенотипические изменения (особенности внешнего вида). При их наличии врач проводит дополнительное обследование для исключения синдрома Патау и других хромосомных аномалий. К характерным симптомам относят:
- долихоцефалические изменения формы черепа. Продольный размер головы преобладает над поперечным;
- уменьшение размера лба при выступающем затылке;
- у детей уменьшены глаза и рот.
Реже встречается кожная складка у внутреннего края глаза, опущение нижнего века, экзофтальм (“выпученные” глаза), косоглазие и укорочение шеи. Деформация ушей представлена уменьшением мочек, отсутствием козелков и низким расположением ушных раковин.
Признаки заболевания включают изменения опорно-двигательного аппарата: деформацию пальцев кистей, уменьшение размеров грудины и ребер, врожденный вывих бедра, косолапость и пр. В связи с нарушением развития тканей у больных появляются папилломы и гемангиомы на кожном покрове.
18 хромосома у человека играет важную роль в формировании внутренних органов. Увеличение числа ее копий или транслокация (изменение расположения) участков приводит к аномалиям их развития. Наиболее часто встречаются дефекты сердечно-сосудистой системы в виде коарктации аорты, тетрады Фалло, изменения положения клапанов и др. Также страдает желудочно-кишечный тракт, мочеполовая система и центральная нервная система. Изменения в строении головного мозга характеризуются гидроцефалией, кистами, гипоплазией отдельных структур и т.п.
Синдром Эдвардса в большинстве случаев приводит к гибели пациентов в первый год жизни. Смерть возникает при отсутствии лечения из-за развития дыхательной или сердечно-сосудистой недостаточности.
Негативные последствия
Нарушения развития внутренних органов и центральной нервной системы приводит к множественным порокам. В связи с аномалиями строения сердца, главных сосудов и органов дыхания у ребенка развивается сердечно-сосудистая или дыхательная недостаточность. Она может стать причиной смерти при отсутствии своевременной медицинской помощи.
Пороки развития органов мочевыделения создают предпосылку для возникновения пиелонефрита, поликистозной почки и других аномалий. В результате их прогрессирования развивается хроническая почечная недостаточность, сопровождающаяся накоплением в крови продуктов азотистого обмена. Это может привести к нарушению работы ЦНС и другим тяжелым последствиям, включая летальный исход.
Последствиями микроцефалии и гипоплазии мозговых структур являются расстройства работы внутренних органов, а также невозможность формирования сложных психических или двигательных навыков. При поражении центров, ответственных за дыхание или сердечно-сосудистую деятельность, развивается их недостаточность.
Диагностика болезни
В диагностике заболевания важно ее внутриутробное выявление. При обнаружении болезни показано искусственное прерывание беременности. Косвенный признак патологии — недоразвитие одной из пупочных артерий, многоводие, уменьшение размеров плаценты и пр. Они могут быть выявлены во время проведения планового УЗИ.
Диагностической ценностью обладают неинвазивные методы, связанные с определением в крови матери маркеров болезни: хорионического гонадотропина, PAPP, альфа-фетопротеина, а также свободного эстрадиола. Изменение уровня указанных веществ в кровотоке женщины является показанием для проведения дополнительных диагностических процедур.
В процессе диагностики заболевания большое значение имеет совокупность факторов: результаты лабораторного скрининга и данных УЗИ, срок гестации и возраст беременной женщины. При наличии нескольких факторов рекомендованы инвазивные исследования — биопсия хориона, амниоцентез (взятие на анализ околоплодных вод) или кордоцентез (взятие анализа крови из пуповины). Полученный в результате процедур материал плода подвергается кариотипированию с подсчетом числа хромосом.
Больные после установления диагноза нуждаются в комплексном обследовании. Рекомендуется провести консультации невролога, кардиохирурга, кардиолога, уролога и других врачей. Всем детям проводят электрокардиографию, ЭхоКГ и ультразвуковое исследование внутренних органов. При наличии неврологического дефицита необходимо снятие ЭЭГ и УЗИ или КТ головного мозга.
Дифференциальную диагностику проводят с различными хромосомными патологиями – синдромом кошачьего крика и пр. В ее основе лежат молекулярно-генетические методы и определение кариотипа.
Подходы к лечению
Хромосомные аномалии не могут быть устранены. В связи с этим проводимое лечение направлено на поддержание жизни больного и устранение имеющихся симптомов. Терапия включает в себя медикаментозные и хирургические методики.
Лекарственные препараты назначаются в следующих случаях:
- наличие инфекционного поражения органов мочевыделительной или дыхательной системы. Применяют антибиотики широкого спектра действия, преимущественно из группы защищенных пенициллинов или цефалоспоринов;
- препараты, улучшающие мозговое кровообращение;
- средства, регулирующие тонус кровеносных сосудов.
Хирургические вмешательства выполняются по поводу аномалий развития сердца и сосудов, органов мочевыделительной системы, желудочно-кишечного тракта. Операции направлены на стабилизацию их состояния и улучшение качества жизни ребенка.
Прогноз для ребенка
Прогноз при хромосомных заболеваниях неблагоприятный. Выздоровление невозможно. Средняя продолжительность жизни — 1-2 года. До возраста 12 месяцев доживает менее 10% больных, а до 10 лет — только 1%. Для мозаичных вариантов синдрома Эдвардса прогноз благоприятнее, что связано с меньшей выраженности аномалий внутренних органов.
Основные причины гибели связаны с декомпенсацией сердечно-сосудистой и дыхательной недостаточности. Летальные состояния развиваются внезапно, несмотря на проводимое лечение.
Возможности профилактики
Профилактика болезни основывается на устранении причин, которые повышают риск развития хромосомных аномалий. К ним относят:
- беременность в возрасте до 30 лет. Чем возраст родителей выше, тем риск возникновения изменений в хромосомах больше;
- избегать работы на вредном химическом и промышленном производстве;
- исключение вредных привычек — табакокурения, употребления алкоголя и наркомании. Указанные воздействия в период беременности и после него способствуют накоплению в половых клетках мутаций, что может привести ко множеству хромосомных аномалий у будущего малыша, измененить его кариотип.
Важным элементом профилактики заболевания является скрининг беременных. Изменения, характерные для синдрома, хорошо видны на УЗИ. Назначение дополнительных инвазивных методик позволяет подтвердить диагноз и прервать беременность на любом сроке.
Развитие синдрома Эдвардса связано с изменениями в хромосомном наборе плода. Заболевание характеризуется неблагоприятным прогнозом, так как большинство детей умирает в первые годы жизни на фоне сердечно-сосудистой или дыхательной недостаточности. Лечение патологии носит симптоматический характер, так как исправление набора хромосом невозможно. Терапия направлена на устранение конкретных симптомов и увеличение продолжительности жизни больного ребенка. В процессе обследования важно исключить другие хромосомные аномалии — синдром Шерешевского-Тернера и пр. Это позволяет уточнить диагноз и прогноз для больного.
Читайте в следующей статье: синдром денди уокера